一文读懂 | 低氧驱动,氧疗,和高碳酸血症
时间:2023-08-06 17:30:36 热度:37.1℃ 作者:网络
要点
● 接受氧疗的COPD急性加重患者PaCO2水平有一定程度的升高。
● 氧疗可使患者的分钟通气量略有下降;然而,PaCO2水平的升高不能单纯由低通气量来解释。
● 此外,分钟通气通量在氧疗数分钟后可恢复到接近基线水平,但PaCO2却持续保持较高水平,提示高碳酸血症的发生还存在其它机制。
● PaCO2水平升高的更可能的机制是氧疗后肺泡内氧分压较高,从而抑制了缺氧性肺血管收缩反应。缺氧性肺血管收缩反应的消失,使得肺内血流再分布,肺通气不良区域的血流增加,从而导致死腔增加和高碳酸血症。
● 根据何尔登效应,血红蛋白在肺内被氧合后,导致其与二氧化碳的结合携带能力下降,从而释放出二氧化碳。然而,因患者通气仍然保持较低水平,不能充分清除二氧化碳,从而引起高碳酸血症。
● 重要的是,不要因为坚信氧疗会抑制“低氧驱动”而产生了非理性恐惧,从而不给缺氧患者进行氧疗。
1 背景
在COPD二氧化碳潴留的患者中,常常需要精细滴定调节氧流量。这一思路背后的驱动原理是一个传统的观念,即氧疗可能会抑制缺氧所致的呼吸驱动,并随着低通气的发生而导致PaCO2水平的升高。甚至有人推断,在这些患者中,缺氧是通过主动脉窦神经活动而形成的一种呼吸刺激。低氧驱动对呼吸衰竭患者,特别是高碳酸血症患者的作用有多重要? 氧疗真的会抑制COPD患者的自主呼吸而引起低通气,并导致CO2水平的显著升高吗? 其他机制(包括死腔的增加和缺氧性肺血管收缩反应的消失)是否更可能诱导高碳酸血症的发生?
2 死腔
2.1 解剖死腔
解剖死腔是指仅作为通道容许空气进入细支气管和肺泡的那一部分气道。在这部分气道内不会发生气体交换。解剖死腔容量大约为150ml。Vd/Vt表示死腔量与潮气量的比例。在低潮气量呼吸时,死腔的相对比例增加(高Vd/Vt),如果分钟通气量不增加,可导致高碳酸血症。
2.2 肺泡死腔
肺泡死腔是指通气的肺泡中血流灌注减少(高V/Q)。因此,肺泡死腔代表了通气充足但血流灌注受损的区域,与肺部通气不良的区域正好相反,那些区域通气减少,血流灌注正常(低V/Q,导致分流)。在大多数肺实质性疾病过程中,高碳酸血症的机制是肺泡死腔的增加。
2.3生理死腔
解剖死腔和肺泡死腔综合起来统称生理死腔。
即:生理死腔=解剖死腔+肺泡死腔
3 氧疗与呼吸驱动:临床证据
Aubier团队分析了20例COPD患者在呼吸衰竭急性期的呼吸模式;对其中12例患者在急性期恢复后做了进一步分析。把结果与相同年龄组的正常受试者进行比较。急性期和慢性期的分钟通气量相似,与正常受试者相当。在急期的呼吸驱动力(用口闭合压力进行衡量)比正常受试者高5倍。氧疗后,口腔闭合压下降了40%,提示呼吸驱动力显著下降。以5 L / min速度氧疗 30 min之后,分钟通气减少14%,主要由于呼吸频率降低,这表明吸气流量减少。氧疗后PaCO2明显升高,但这升高的PaCO2与分钟通气量的减少并不相称,所以这提示着高碳酸血症的发生可能还有其它机制。作者认为,死腔增加可以解释氧疗之后发生的高碳酸血症。
在后来的一项研究中,Aubier团队纳入了22例急性感染加重期的COPD患者,对其从吸入室内空气转为吸入100%氧气后的通气量和动脉血气变化情况进行评估。患者在呼吸室内空气时,均存在缺氧(平均PaO2 38±2 mmHg)和高碳酸血症(PaCO2 65±3 mmHg)。在吸入100%氧气15分钟期间,对呼吸参数进行监测。给予100%氧气后,观察到分钟通气开始下降,最低值出现在吸入纯氧20~180s之间。在开始吸入100%的氧气后,分钟通气量开始减少,随后分钟通气量逐渐增加,并在大约12分钟后达到平台期。在15分钟的研究期结束时,分钟通气的平台水平是基线水平的93%±6%。然而,PaCO2在整个研究期间持续升高,平均升高23±5 mmHg(图1)。PaO2从平均基线水平38±2 mmHg升高到225±23 mmHg。吸入100%氧气时,潮气量和呼吸频率无明显变化。PaCO2的升高不成比例,这不能用分钟通气量的微小下降来解释。在COPD急性加重期,通常可观察到呼吸驱动的显著增强。上述研究表明,尽管氧疗后呼吸驱动可能略有下降,但仍高于正常。仅仅呼吸驱动的下降不能解释PaCO2水平的升高。

图1.在COPD急性加重期,从吸入空气转为吸入100%氧气的影响。开始时分钟通气量减少,随后逐步上升达到一个平台期;然而,PaCO2持续升高,提示高碳酸血症的发生还有其机制存在。
4 何尔登效应
血红蛋白以氨基甲酰化合物的形式携带二氧化碳。根据何尔登效应,当氧合血红蛋白水平升高时,二氧化碳解离曲线向右移。因此,氧合血红蛋白携带的二氧化碳较少。在正常肺中,当血红蛋白被氧合成氧合血红蛋白时,二氧化碳被释放出来并呼出去。以COPD患者为例。氧疗后可如预期那样使血红蛋白更易释放出结合的二氧化碳。然而,由于COPD患者通气量没有相应增加,血红蛋白释放出的二氧化碳得不到充分呼出。这导致了PaCO2水平的升高。
5 缺氧性肺血管收缩反应的消失
当肺泡内氧分压下降时,肺循环的平滑肌收缩,导致血管收缩。这是在缺氧的情况下,降低肺内分流的反射机制。肺动脉对缺氧的血管收缩反应与在体循环中发生的缺氧诱导的血管扩张作用正好相反。氧疗时,肺泡氧分压升高。氧水平的升高导致缺氧性肺血管收缩反应被抑制,使得血流从通气较好的肺区再分布到通气较差的肺区,局部的V/Q比值减少。相反,在通气较好的肺部区域,由于血流被分流,使得血供相对于通气出现了减少,导致较高的V/Q比值和死腔通气增加。Aubier团队在他们的研究中也观察到了死腔的显著增加,给予100%氧气15分钟后,死腔通气量从77±2增加到82±2。上述两种机制都可能导致PaCO2水平升高。Robinson团队在22例COPD急性加重的患者当中评估了氧诱导的高碳酸血症的机制。患者先是呼吸空气,然后通过鼻罩吸入100%氧气至少20分钟。在呼吸空气和100%氧时均对通气、V/Q比值和心输出量进行监测。如果PaCO2升高超过3 mmHg,则分类为二氧化碳潴留者;如果PCO2升高小于3 mmHg,则分类为非二氧化碳潴留者。二氧化碳潴留组患者有12例,PCO2水平升高了8.3±5.6 mmHg;非二氧化碳潴留组,PCO2水平变化为−1.3 ± 2.2 mmHg。吸入100%氧气20分钟后,二氧化碳潴留组的分钟通气量由9.0±1.5L/min降至7.2±1.2L/min。然而,CO2的增加似乎与分钟通气量的减少并不成比例。两组的V/Q不匹配程度相同,可能是由于缺氧性肺血管收缩反应被抑制。二氧化碳潴留组的死腔通气显著增加,这可能导致CO2升高。
Hanson团队把来自肺循环模拟计算机模型的数据与一系列COPD患者的数据进行了比较。模拟氧疗模型产生的数据与接受氧疗的COPD患者的数据相似。作者观察到,氧疗导致生理死腔的增加,是由缺氧性肺血管收缩反应被抑制以及何尔登效应所致。他们认为,仅生理死腔的增加,就足以解释接受氧疗的患者PaCO2升高的原因。图2描述的是氧疗后PaCO2升高的机制。
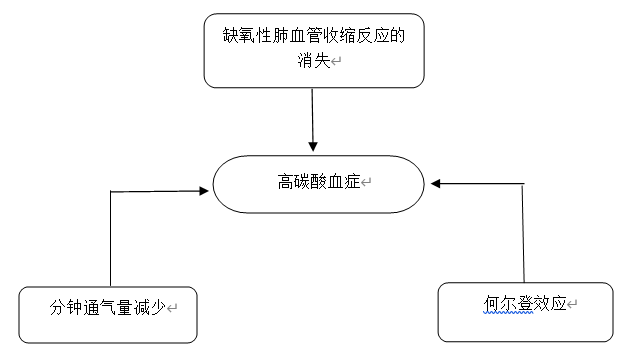
图2. 在接受氧疗的COPD急性加重期患者中观察到的高碳酸血症的潜在机制。

