Nature:剑桥大学徐宇/Michael Inouye创建并验证与应用大规模的多组学特征基因评分模型
时间:2023-03-31 14:31:18 热度:37.1℃ 作者:网络
北京时间2023年3月29日,英国剑桥大学Michael Inouye团队(徐宇为第一作者和共同通信作者)在Nature发表了一篇题为“An atlas of genetic scores to predict multi-omic traits”的文章。
该研究创建、验证、应用了大规模的多组学特征(multi-omic traits)基因评分(genetic score)模型,揭示了一系列新的疾病生物学机理,并为该领域开发了一个公开资源平台,为多组学特征基因评分的广泛应用提供了重要基础。

多组学分析是指综合不同组学领域的知识(如基因组学、转录组学、蛋白质组学和代谢组学),对多个层次的生物分子特征进行量化和分析,以全面理解生物系统中的分子关系和功能。它能帮助我们深入理解生物过程,发现新的治疗靶点和生物标志物,为疾病预防、诊断和新型药物开发提供关键的科学支撑。
然而,由于多组学数据的收集和获取成本极高且耗时,大规模人群队列通常只为少部分参与者(或不为任何参与者)产生多组学数据,这极大地降低了研究结果的可靠性与准确性,甚至可能限制相关重要科研工作的开展。
多组学特征通常是可遗传的。因此,基因组信息可以用来预测其他多组学分子特征水平,例如转录、蛋白质、代谢产物和其他生物分子等。利用基因预测的组学特征,我们可以在大规模队列中经济高效地开展多组学分析,深入探究目标疾病的致病机理。
该研究利用机器学习方法及一个涵盖有丰富多组学数据的健康献血者队列(N = ~50000参与者),为逾17000个多组学特征建立了基因评分(genetic score)模型,其中包括2692 蛋白质, 867代谢产物及13668转录产物。研究揭示了大量组学分子特征在遗传上具有高度可预测性,例如其中409个基因评分模型对训练数据的解释性(R2)超过了50%。
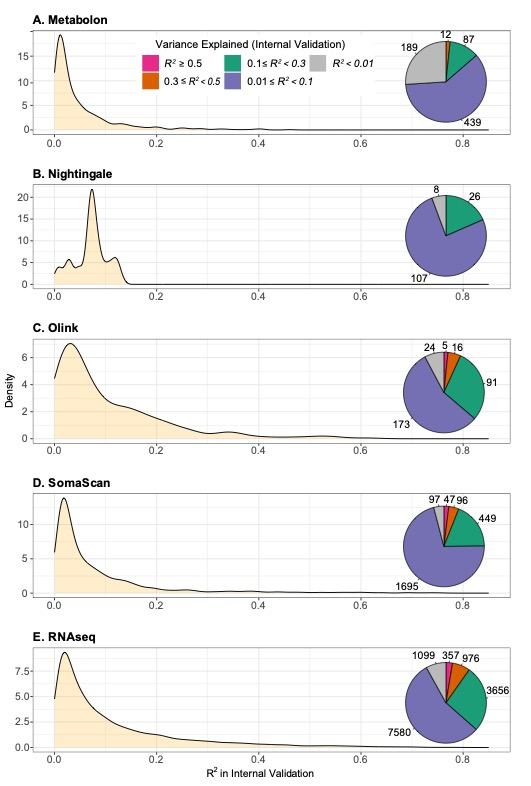
图1. 多组学基因评分在训练数据中的预测准确度。饼状图反映了在特定R2范围内的基因评分数量
该研究进一步在涵盖多个种族的外部人群队列中评估了所建立基因评分模型的准确性,其中包括欧洲、东亚、南亚和非洲裔美国人在内的七个人群队列。研究发现了大多数基因评分模型在同源欧洲人群中的预测准确度与在训练数据(欧洲人群)中高度一致,同时揭示了其预测准确性在非欧洲人群中明显降低。
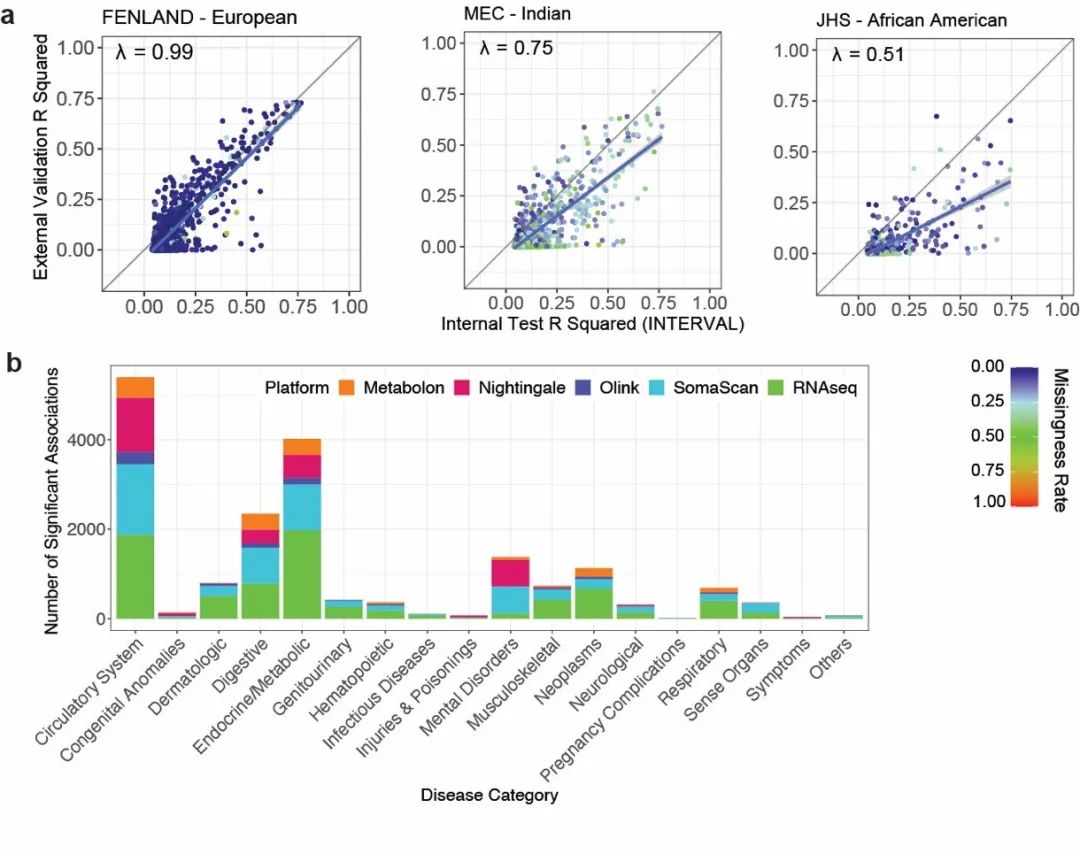
图2. 多组学基因评分模型的验证与应用。a. SomaScan蛋白质基因评分模型在训练数据与外部队列中的预测准确度比较。b. 利用多组学基因评分模型在UK Biobank上的全表型组关联分析。柱状图反应了该分析所检测到的多组学特征与各类疾病之间的关联数量
该研究利用所建立的基因评分模型评估了生物通路受基因控制的程度,并揭示了受基因控制较大的生物通路,例如消化和吸收(digestion and absorption)通路。这帮助我们更深入地理解了疾病的可遗传性及其分子遗传机制。
该研究利用所建立的基因评分模型为UK Biobank中近50万名参与者生成了多组学数据,并通过全表型组关联分析(phenome-wide association analysis, PheWAS),发现了大量多组学特征与复杂疾病之间的关联,并揭示了一系列新的遗传机制和病理生理学知识(例如JAK-STAT信号通路与冠状动脉疾病之间的关联)。
该研究开发了一个公共资源平台(OmicsPred.org),并在该平台中开源了所建立的多组学特征基因评分模型,及相关验证与应用结果。同时,OmicsPred还将作为一个中央资源平台,为其他科研工作者分享与开源他们开发的多组学特征基因评分模型及相关结果提供平台支撑。
原始出处:
Xu, Y., Ritchie, S.C., Liang, Y. et al. An atlas of genetic scores to predict multi-omic traits. Nature (2023). https://doi.org/10.1038/s41586-023-05844-9.

