文献荐读|胡兴胜教授:由IGF2BP3驱动的代谢重编程促进非小细胞肺癌对EGFR-TKIs的获得性耐药
时间:2023-10-31 23:43:08 热度:37.1℃ 作者:网络
01 研究背景
肺癌是癌症相关死亡的主要原因,其中非小细胞肺癌(NSCLC)约占85%。自1997年以来,从第一代厄洛替尼到第三代奥希替尼,EGFR酪氨酸激酶抑制剂(TKI)在非小细胞肺癌的靶向治疗中具有显著的临床疗效。不可避免的是,越来越多的患者对靶向治疗产生耐药性,这对TKI治疗是一个巨大的挑战。因此,目前迫切需要探索潜在的耐药机制并制定有效的策略来克服TKI获得性耐药的发生。
代谢适应是肿瘤为满足恶性细胞对生物能量、生物合成和解毒不断增加的需求而存在的。代谢重编程指肿瘤的代谢特性和偏好在肿瘤进展中发生改变,这可能是耐药的发生机制。与经典的Warburg效应相反,Sarry等人观察到阿糖胞苷耐药的AML细胞和患者表现出氧化磷酸化(OXPHOS)活性升高,OXPHOS抑制剂可以使耐药AML细胞对阿糖胞苷敏感。然而,目前尚不清楚NSCLC细胞是否可以通过代谢适应来重建其微环境,从而产生耐药。
人胰岛素样生长因子2 mRNA结合蛋白家族(IMP2/IGF2bp)是一类包含2个N端RNA识别结合区(RRM)和4个C端异源核糖核蛋白KH结构域的胚胎蛋白,可结合目标mRNA编码区域,调控RNA转运、翻译和降解,并进一步参与发育、肿瘤发生和干细胞形成等一系列生物学过程。近期研究表明IGF2BP3家族是N6-腺苷酸甲基化阅读蛋白,KH结构域为识别m6A GC序列的核心结构域,可促进下游靶基因mRNA的稳定和翻译。近年来,多项研究发现基因的转录后调控因子IGF2BP3参与肿瘤细胞的增殖、侵袭和化疗耐受。IGF2BP3在多种肿瘤中表达上调,并与患者预后不良相关。然而,其具体作用机制和潜在的功能尚不清楚。
02 研究对象及方法
通过对原代组织样本和细胞系培养、构建质粒、转染细胞和病毒感染等操作构建了敲除和过表达细胞系,并对稳转细胞系进行了Western-blot分析、qRT-PCR检测、细胞活力测定、细胞增殖试验、克隆形成试验和微球形成试验、细胞凋亡分析、RNA测序、RNA免疫沉淀测序、Seahorse检测、荧光素酶报告基因试验和诱变试验、m6A-RNA免疫沉淀试验(RIP)等,并对细胞的ROS、ATP、线粒体膜电位水平(MMP)、代谢组学、NAD+/NADH、RNA和蛋白质的稳定性进行了测定。此外,作者利用BALB/c裸鼠构建了Dox-shIGF2BP3皮下肿瘤模型和IGF2BP3过表达皮下肿瘤模型,并进行了IGF2BP3皮下肿瘤模型的拯救实验。最后,作者构建了NSCLC患者来源的类器官模型(PDO)并进行活性测定。
03 结果
一 IGF2BP3在获得性EGFR-TKI耐药的NSCLC中上调
对比长期接受TKI治疗的获得性耐药NSCLC肿瘤和TKI敏感NSCLC肿瘤的转录组数据,并对比TKI耐药NSCLC样本和HCC827细胞转录组数据的DEGs和三个独立临床GEO队列数据库的DEGs,发现与NSCLC获得性TKI耐药相关的4个上调基因和3个下调基因,IGF2BP3是其中一个上调基因,与多种肿瘤耐药相关。7种NSCLC细胞系IGF2BP3蛋白表达均上调。免疫组化显示IGF2BP3在NSCLC组织中显著上调。与正常组织或TKI敏感样本相比,获得性TKI耐药样本IGF2BP3 mRNA和蛋白显著升高,细胞试验结果相同。以上结果均支持IGF2BP3介导NSCLC TKI获得性耐药的发生。
二 IGF2BP3促进NSCLC获得性EGFR-TKI耐药发生
IGF2BP3敲除使得敏感和耐药NSCLC细胞系对吉非替尼的敏感性显著增加。吉非替尼可显著降低IGF2BP3耗竭NSCLC细胞系的细胞增殖、克隆形成和微球形成能力。流式细胞术结果也显示IGF2BP3耗竭导致细胞凋亡显著增加,对吉非替尼的敏感性增强。相反,IGF2BP3过表达使得两种吉非替尼敏感NSCLC细胞系的敏感性降低。小鼠模型中,IGF2BP3敲除细胞系皮下移植瘤体积和质量较小,对吉非替尼的敏感性较高,而IGF2BP3过表达皮下移植瘤生长迅速,对吉非替尼的敏感性较低。(下图)
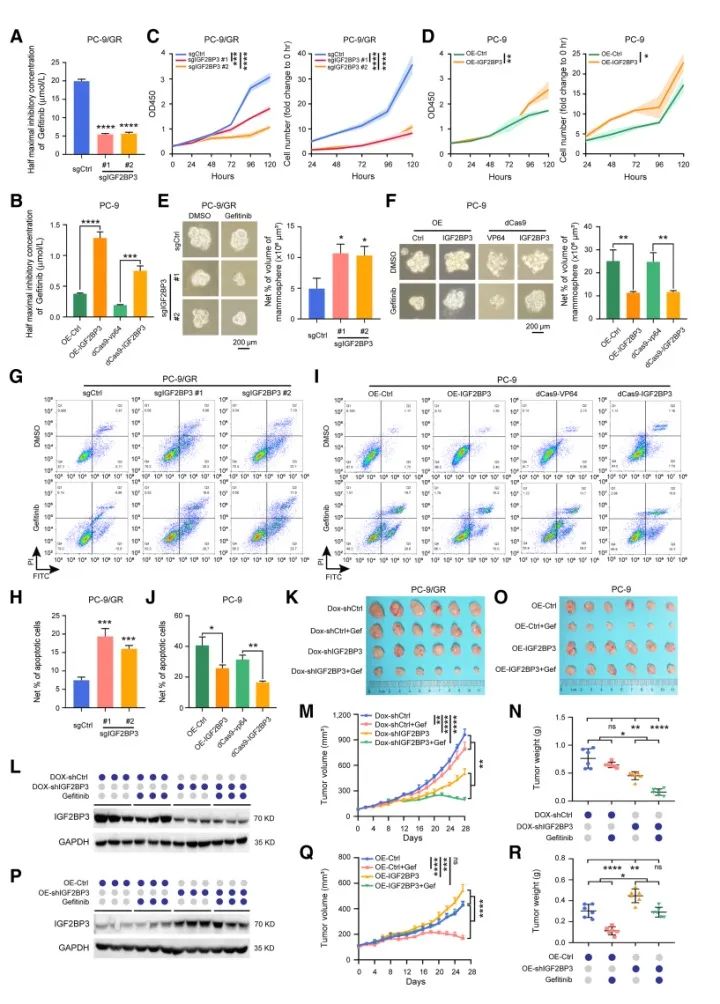
三 在获得性EGFR-TKI耐药的NSCLC中,IGF2BP3通过COX6B2介导线粒体能量重编程
生物能量分析表明,与TKI敏感细胞系相比,TKI耐药细胞的胞外酸化率(ECAR)略有下降,线粒体氧耗率(OCR)高出近2至3倍,表明其存在高水平OXPHOS。此外,线粒体OXPHOS复合体、总ROS、线粒体ROS、ATP和MMP水平在TKI耐药细胞系中均显著上调,均提示TKI耐药细胞系拥有更高的OXPHOS能力,可产生大量ATP用于供能。COX6B2敲除细胞系OXPHOS水平显著降低,ROS生成和ATP生成显著减少。IGF2BP3稳定敲低细胞系的OXPHOS能力明显降低,而进一步过表达COX6B2可明显改变这种情况。PC-9/GR和PC-9细胞系中,总ROS、线粒体ROS、ATP和MMP水平与IGF2BP3表达呈正相关, IGF2BP3的敲低可被COX6B2过表达逆转。综上,IGF2BP3-COX6B2轴在TKI耐药细胞系的代谢重编程中起关键作用。(下图)
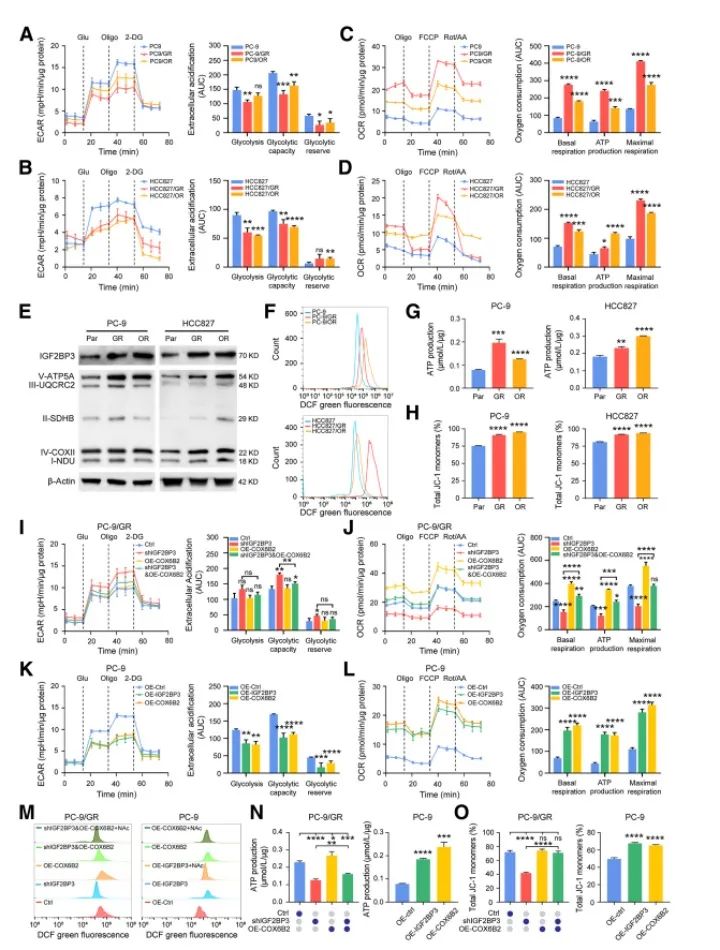
四 IGF2BP3以依赖m6A的方式调控COX6B2的翻译
与TKI敏感细胞系相比,TKI耐药细胞系COX6B2表达上调,两种细胞系中COX6B2和IGF2BP3蛋白水平均呈正相关。IGF2BP3敲低细胞的COX6B2 mRNA和蛋白半衰期显著降低,而IGF2BP3过表达细胞的COX6B2半衰期显著增加,表明IGF2BP3可增加NSCLC中COX6B2 mRNA的稳定性。此外,研究发现m6A修饰缺陷细胞系中COX6B2下调,半衰期显著降低,而IGF2BP3保持不变,给予外源野生型m6A修饰的细胞可逆转这一改变,表明IGF2BP3可能以依赖m6A的方式调控COX6B2的翻译。对稳定过表达IGF2BP3的细胞系进行IGF2BP3-RIP和m6A-RIP检测,发现IGF2BP3与COX6B2 mRNA的结合以及COX6B2 mRNA的m6A水平均显著升高。上述结果表明,COX6B2 mRNA的m6A修饰是IGF2BP3结合和诱导NSCLC细胞耐药的必要条件。(下图)
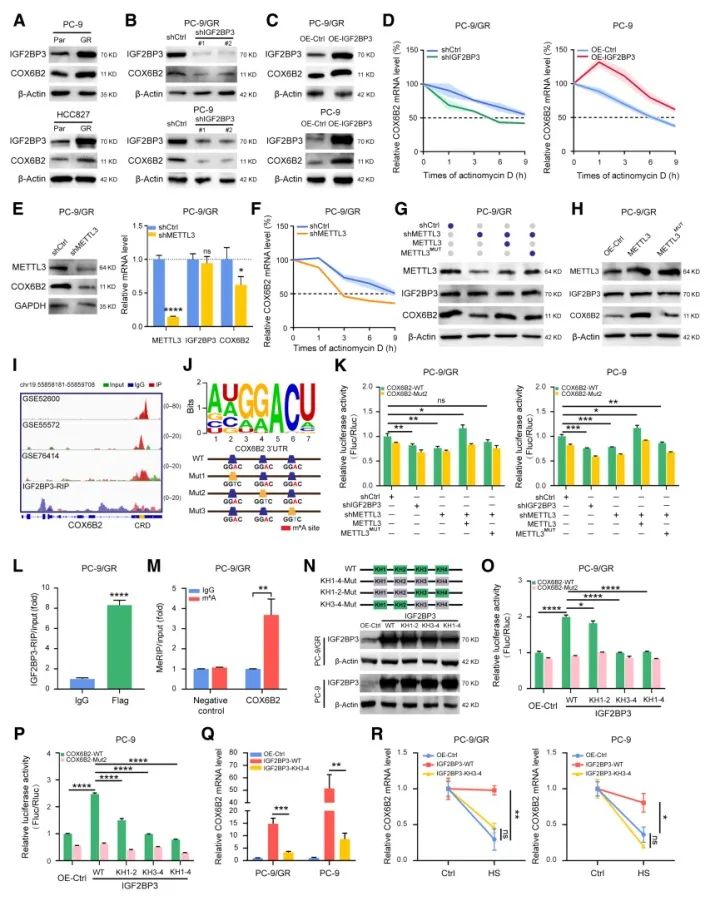
五 OXPHOS抑制剂可增强EGFR-TKIs的疗效
小分子OXPHOS抑制剂IACS-010759可显著增加耐药细胞对吉非替尼的敏感性。同样,IACS-010759联合吉非替尼处理TKI耐药细胞系可诱导凋亡,抑制增殖和集落形成。裸鼠PC-9/GR细胞建立的CDX和PDX模型结果同体外试验一致,IACS-010759联合吉非替尼可显著抑制耐药CDX/PDX的生长,且未观察到明显的毒副反应。上述结果从多方面证明了OXPHOS抑制剂在TKI耐药模型中的作用,提示OXPHOS抑制剂具有恢复NSCLC患者对TKI敏感性的潜力。
04 结论
靶向IGF2BP3可以显著增强NSCLC对TKIs的敏感性,其效果与IGF2BP3在线粒体OXPHOS能量传递中的关键下游靶点COX6B2的协调显著相关。从机制上讲,IGF2BP3可以m6A依赖的方式结合到COX6B2的3'非翻译区来增加COX6B2 mRNA的稳定性,从而增加氧化磷酸化、调节烟酰胺代谢,最终驱动肺癌对EGFR-TKI产生治疗抵抗。此外,IACS-010759 对吉非替尼耐药的PDX模型产生强烈的生长抑制作用,表明OXPHOS抑制剂对TKI耐药的NSCLC具有治疗潜力。总而言之,该研究揭示了NSCLC TKI获得性耐药的新机制,即IGF2BP3-COX6B2轴介导的表观遗传修饰和代谢重编程之间依赖IGF2BP3的交互作用,提供了通过靶向代谢改变来克服获得性耐药的新思路。
主编评语
该研究报告了代谢重编程相关分子机制在驱动NSCLC获得性EGFR-TKI耐药中的关键作用。基于生物测定、转录组测序和患者样本信息,作者首次证明了IGF2BP3在EGFR-TKI耐药的NSCLC中上调,IGF2BP3的高表达与较差的临床预后和较高的病理分期显著相关,进一步明确了IGF2BP3的致癌作用。此外,IGF2BP3敲除NSCLC细胞系的增殖和球化能力下降,凋亡增加,对吉非替尼的敏感性提高。IGF1R抑制剂利西替尼联合吉非替尼可有效提高NSCLC的疗效,提示靶向IGF2BP3可能是克服NSCLC获得性耐药的有效策略。
为了阐明IGF2BP3介导NSCLC TKI耐药的分子机制,作者在富集分析的基础上,将重点放在与人类癌症耐药高度相关的代谢途径上,确定了IGF2BP3作为下游靶点。当糖酵解被抑制时,肿瘤可激活其他替代途径来维持生存。生物能量学分析表明,与敏感细胞相比,耐药NSCLC细胞系OXPHOS、ROS、ATP和MMP水平显著提高,表明代谢重编程是耐药肿瘤的潜在治疗靶点。此外,该研究发现COX6B2的存在可有效缓解IGF2BP3耗竭引起的代谢紊乱,证实IGF2BP3-COX6B2轴在TKI耐药的NSCLC代谢重编程中起关键作用,氧化还原酶COX6B2可作为潜在候选靶点。最后,作者确定依赖m6A修饰的COX6B2是IGF2BP3的直接下游靶点,并通过代谢组学分析确定烟酰胺可通过调节细胞内NAD水平驱动TKI耐药NSCLC的OXPHOS代谢。
总之,该研究发现IGF2BP3在TKI耐药NSCLC中以m6A依赖的方式发挥重要作用,IGF2BP3-COX6B2轴是TKI耐药的关键调节因子,并与表观遗传修饰和线粒体功能相关。重要的是,代谢性重编程在TKI耐药NSCLC中起着至关重要的作用,OXPHOS抑制剂是克服NSCLC获得性耐药的潜在治疗选择。

