遗传代谢性疾病概述、髓鞘形成不良丨影像诊断要点
时间:2022-11-06 11:59:26 热度:37.1℃ 作者:网络
遗传代谢性疾病(inherited metabolic disorders,IMD) 很难诊断:疾病可发生于任何年龄;症状依据发病年龄和生化缺陷的严重程度而不同。轻症患者和重症患者相比,大脑的受累部位可能完全不同。随着人体从婴儿期到童年期再到成年期,在大脑的不同部位,相同的酶可能有不同的功能。无疑,若大脑的受累部位不同,患者将会表现出不同的症状。
许多疾病的影像学表现相互重叠,经常随着疾病分期的不同和疾病的变异而不同。疾病早期的影像学是最有帮助的。从神经影像学的角度来说,最有效的方法是,根据疾病早期的MR脑部受累方式来分类疾病。这种方式可通过代谢性数据(通过质谱分析获取)、弥散数据(通过弥散张量成像获取)和偶尔通过磁化传递进行补充。

 正常变异
正常变异
髓鞘形成不良
概述
-
相对于年龄,白质髓鞘形成的减少或缺失程度
-
可能是原发性髓鞘形成不良综合征或继发于其他病理过程
影像
最佳诊断要点
-
>1岁的儿童,T1WI成像上灰白质分界不清
-
>2岁的儿童,T2WI成像上灰白质分界不清
-
部位:评估的关键区域是内囊、锥体束和外周的额叶白质分支
-
大小:髓鞘形成不良将会导致脑体积的减小;在矢状位成像上,胼胝体明显变薄
-
形态:通常形态正常
-
有髓鞘的白质在T1WI上呈高信号
-
短T1反映存在含有脂质蛋白的成熟少突神经胶质细胞
-
T1WI成像显示髓鞘形成完成需要一年时间
-
有髓鞘的白质在T2WI上呈低信号
-
短T2反映由髓鞘包裹的轴突的间质水的移位
-
T2WI成像显示髓鞘形成完成需要3年时间,通常为2年时间
主要的鉴别诊断
-
Pelizaeus-Merzbacher病、18q综合征
-
粘多糖贮积症、线粒体脑病
-
脑白质营养不良症、毛发低硫营养不良
病理
-
髓鞘形成不良通常反映含蛋白脂质蛋白质(PLP) 或碱性髓鞘蛋白(MBP) 异常
-
PLP的缺陷妨碍了正常髓鞘的压紧
-
MBP的作用被认为是在主致密线上稳定髓鞘的螺旋盘绕
诊断纲要
-
区别髓鞘形成不良、髓鞘形成障碍和脱髓鞘是不可能的
-
根据年龄来定义髓鞘的发育程度是可行的:“髓鞘发育的程度相当于X个月的年龄”
-
评价髓鞘形成优先于知晓病人的生理年龄
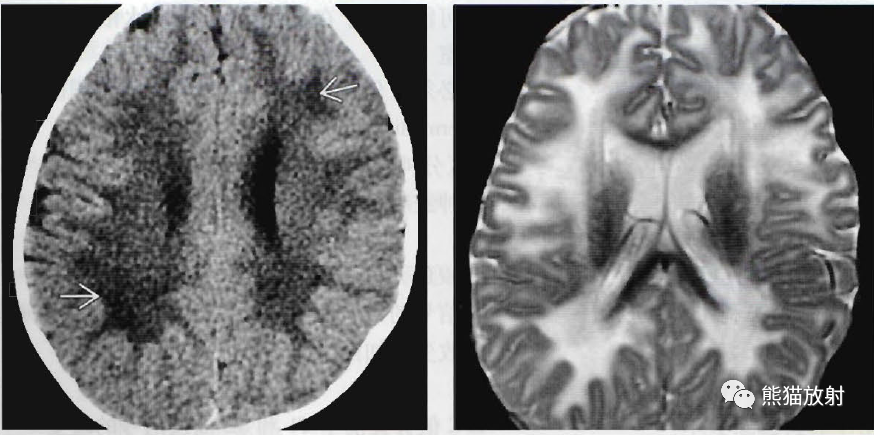
(左) 患有Jacobsen综合征(11q染色体缺失)的一周岁儿童, 轴位NECT显示不规则的低密度,皮层下脑白质最为严重;
(右)同一位患者,轴位T2WI证实CT上的密度减低符合显著的、弥散性髓鞘形成不良。尽管发病率很难确定,在为数众多的染色体缺失综合征中存在髓鞘形成不良。
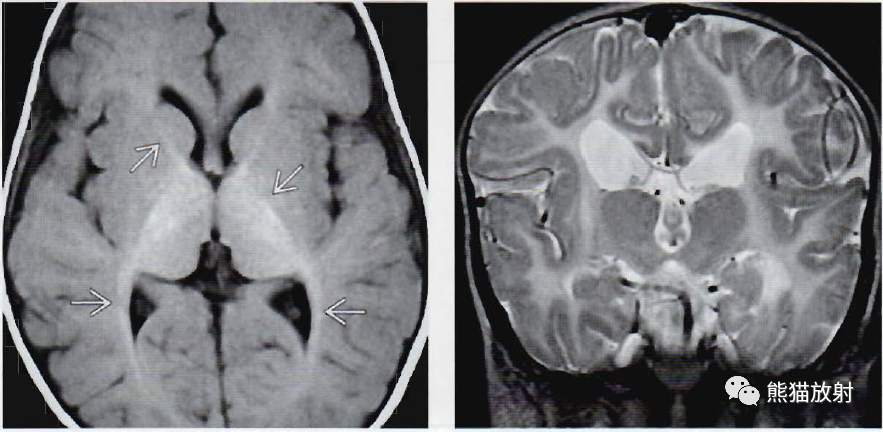
(左)9个月的18q综合征婴儿,轴位T1WI显示微弱的短T1信号,局限于内囊和视辐射。在9个月大的婴儿,在T1WI像上只有白质的大部分远端分支不呈高亮信号。
(右) 一位髓鞘形成不良、基底节和小脑萎缩(H-ABC) 的14岁儿童的冠状位T2WI成像。注意尾状核和胼胝体缺失,另外可见无髓鞘的白质呈异常的高信号。
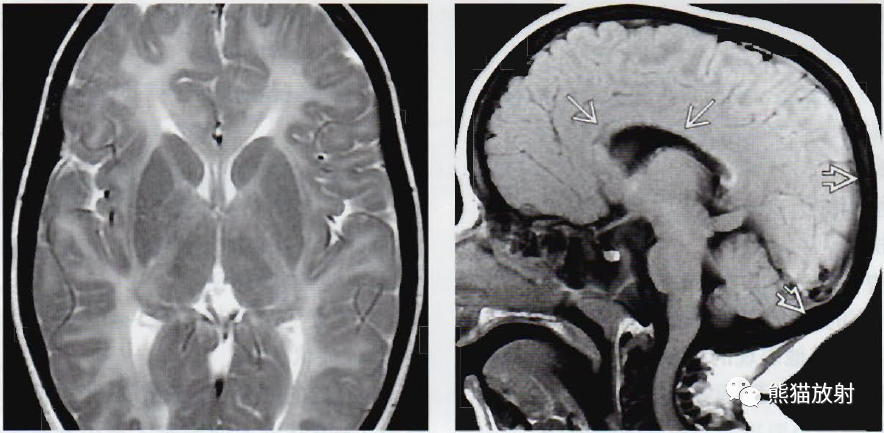
(左) 毛发低硫营养不良的12岁儿童,轴位T2WI显示遍及整个大脑的几乎完全性的髓鞘形成缺失。头发的偏振光下检查显示特征性的“虎-尾”样条带, X线检查显示中心性骨硬化。
(右) 同一位患儿,矢状位T1WI显示遍及大脑的髓鞘沉积的缺失,使得胼胝体在正中位成像上很难清楚地显示其轮廓。注意后部的颅骨增厚。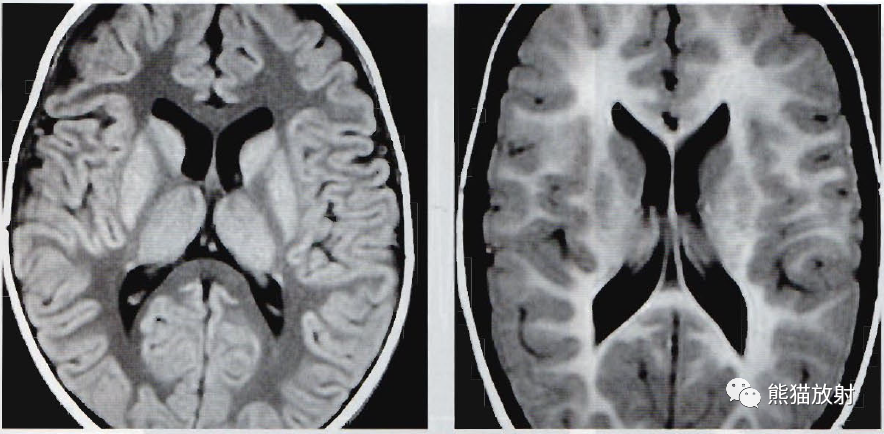
(左)眼球震颤和前向性步态蹒跚的26个月儿童,诊断为Pelizaeus-Merzbacher病, 图像为轴位T1WI成像。均质性的髓鞘沉积缺失所引起的表现类似于成熟大脑的正常FLAIR成像。
(右) 相反,Pelizaeus-Merzbacher病的6岁儿童,轴位FLAIR成像表现类似于正常的T1WI成像,相对于灰质,全部呈高信号。
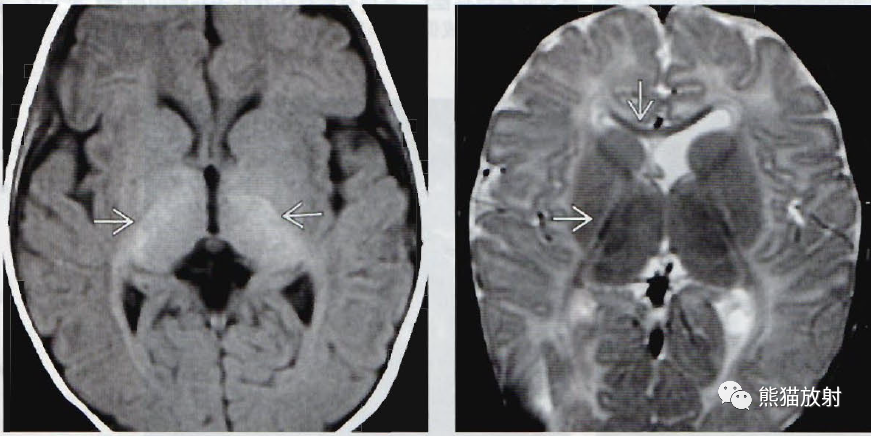
(左)眼球震颤的2岁儿童, 轴位T1WI显示内囊后肢(PLIC) 呈轻度短T1信号;在这个年龄的T1WI成像上,应该表现为髓鞘完全形成。染色体分析显示PLP基因的突变,证实了Pelizaeus-Merzbacher病的诊断。
(右)动静脉瘘的14个月婴儿,轴位T2WIMR显示仅在膝状体和内囊区存在髓鞘成熟的低信号。伴随疾病是髓鞘成熟延迟的一个常见病因。

